Muscular Dystrophy (towards a cure).
Now the gene for DMD/BMD and some of the other MDs have been
isolated and sequences, and we know their approximate functions. How can we design rational strategies to halt
and perhaps even reverse the course of these diseases? Ideally, we would wish
to have a good animal model as the early trials may well be uncomfortable. However, we may have to do without
attempting instead to go straight to clinical trials with human volunteers.
The MDX mouse, is
it a sufficient model for DMD/BMD? Despite the fact that the MDX mouse
lacks dystrophin it seems remarkably unaffected. MDX mice have similar life span expectancy to wild type mice
(around 2 years), and do not show any clinical signs. The two papers given at the end of the first dystrophin lecture
(Grady et al 1997 and Deconinck et al, 1997) conclude that both utrophin and
dystrophin have to be ablated in mice in order to recreate the pathologies seen
in DMD/BMD patients who of course only have deficiencies in the dystrophin
gene. This is a major . Although these and other experiments show
that the MDX mouse is not a good model for the human disease and that the
double K/O mice are also not ideal, they show that utrophin can substitute for
dystrophin at least in the mouse. This
at least is encouraging!

Can dystrophin be introduced to the muscles
of DMD patients? The introduction of dystrophin into the muscles of
patients who lack parts of or the whole of dystrophin is very likely to result
in the patient producing antibodies to this protein as it will be perceived as
being “foreign”. It may be possible to
damp down the patients immune system in the same way that is routine after
transplantation surgery, but this is not ideal. Another problem is that the cDNA of the full length dystrophin
would be very long, a way around this is to try the “mini dystrophin” cDNA
based on the gene product that a very mild BMD patient expressed (England et
al, ).
Figure 10 Utrophin is structurally similar to
dystrophin, but is two helical
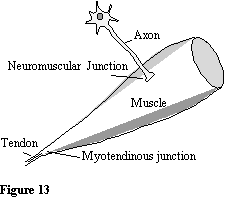
Figure 11. Utrophin is expressed at the myotendinous junction and the
neuromuscular junction in adult human.
repeats shorter
Figure 12

 Utrophin,
a substitute for dystrophin. As utrophin is so similar in structure and
function to dystrophin many groups have concluded that some of the symptoms of
DMD/BMD may be reduced if one were able to persuade muscle cells to produce
utrophin. The studies with the MDX mice
quite clearly show that utrophin does compensate for the lack of dystrophin in
the mouse. Does this necessarily mean
that the same would follow for humans?
There are indications that this is so.
It has frequently been noted that even in the most severely affected DMD
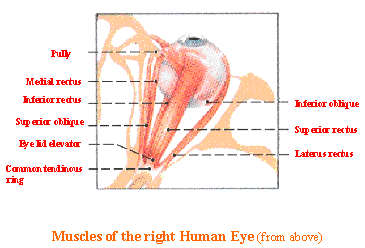
patient that the extraocular muscles (around the eye, See Fig 12) are spared
(Karpati & Carpenter, 1986). These muscles are rather special in that they
express unusual myosin II isoforms and in that they express utrophin in
addition to dystrophin. It seems that the extraoccular muscles are devoid of
DMD pathologies because of the presence of utrophin as mice lacking utrophin
and dystrophin show degenerationeven in the extraocular muscles (Porter et al,
1998). Usually, utrophin expression in skeletal muscles is restricted to the
myotendinous junction and the neuromuscular junction (Fig 11). This is also the case in mouse, but here
utrophin is found in skeletal muscle in the
embryo, and in the MDX mouse where muscle regenerates. It is tempting to speculate that if one
could artificially express utrophin in the muscle of DMD/BMD patients that this
may substitute for dystrophin to some extend and alleviate the symptoms of the
disease.
Utrophin,
a substitute for dystrophin. As utrophin is so similar in structure and
function to dystrophin many groups have concluded that some of the symptoms of
DMD/BMD may be reduced if one were able to persuade muscle cells to produce
utrophin. The studies with the MDX mice
quite clearly show that utrophin does compensate for the lack of dystrophin in
the mouse. Does this necessarily mean
that the same would follow for humans?
There are indications that this is so.
It has frequently been noted that even in the most severely affected DMD
patient that the extraocular muscles (around the eye, See Fig 12) are spared
(Karpati & Carpenter, 1986). These muscles are rather special in that they
express unusual myosin II isoforms and in that they express utrophin in
addition to dystrophin. It seems that the extraoccular muscles are devoid of
DMD pathologies because of the presence of utrophin as mice lacking utrophin
and dystrophin show degenerationeven in the extraocular muscles (Porter et al,
1998). Usually, utrophin expression in skeletal muscles is restricted to the
myotendinous junction and the neuromuscular junction (Fig 11). This is also the case in mouse, but here
utrophin is found in skeletal muscle in the
embryo, and in the MDX mouse where muscle regenerates. It is tempting to speculate that if one
could artificially express utrophin in the muscle of DMD/BMD patients that this
may substitute for dystrophin to some extend and alleviate the symptoms of the
disease.



 How do you get
Utrophin cDNAs into muscle cells?
Muscle cells have an usual ability to take up naked DNA and shuttle it
into their nuclei. This of course led
some to experiment with the simple injection of dystrophin cDNA constructs
directly into muscle. However, the
earlier optimism was curtailed as it soon became clear that the efficiency of
this process was nowhere near sufficient.
How do you get
Utrophin cDNAs into muscle cells?
Muscle cells have an usual ability to take up naked DNA and shuttle it
into their nuclei. This of course led
some to experiment with the simple injection of dystrophin cDNA constructs
directly into muscle. However, the
earlier optimism was curtailed as it soon became clear that the efficiency of
this process was nowhere near sufficient.
Figure 13. The use of the
"adeno-associated" virus (AAV) to introduce a working copy of the
sarcoglycn gene to the foot muscle of a LGMD patient as an early trial of
the technique. (adapted from the MDA web site http://mdausa.org/home.html )
The first human trial for a
genetic treatment of a Muscular Dystrophy.
The first treatment for LGMD was started on the 2nd Sept
1999. Donavon Decker, a 36-year-old air traffic controller became the first
person to receive a gene therapy injection for has a sacroglycan deficiency
LGMD. The sacroglycan cDNA was inserted
in a construct containing muscle-specific promotors in a "adeno-associated"
virus (AAV) injected a muscle on the top of the foot. AAV is not pathogenic for humans but the replicative machinery
was removed in the strain used for this study to be safe. The AAV carrying the sarcoglycan cDNA was
injected into a muscle in one foot while the same muscle of the other foot
received a sham injection. Biopsies will be taken from the muscle of both feet
to compare the condition of the two muscles after six weeks. Integration of the viral DNA into the
genomic DNA will be checked in addition to expression of the sarcoglycan
construct. Evidence for the appearance
of the previously absent sacroglycan (and the other sacroglycans, whose
expression is also dramatically reduced in this LGMD) will be monitored. Damage
to the muscle cells will also be investigated.
The patient will be monitored closely for any adverse reactions,
including immune response or other unwanted response to the virus used to carry
the genes, to the genes themselves, or to the protein molecules produced by the
genes. In anticipation of all going
well with this single patient trial the MDA are gearing up for a full scale
trial. The results of the trial will be
published by the MDA on their web-site at:- http://mdausa.org/home.html
Figure 11
Although the AAV is known not to
cause serious immune response it is not certain whether this comparatively
massive dose will cause some problems.
If the sacroglycan cDNA is expressed in the muscle, it is possible that
the pateints immune system will see this as foreign. There are problems to be overcome, but this is clearly a
promising development.
Can the same process work for DMD/BMD?
The
main difference between DMD/BMD and sacroglycan LGMD is size. The dystrophin cDNA is huge and requires a
different viral delivery system than that being used in the LGMD trials.
Unfortunately, the best available virus large enough to contain the dystrophin
gene, the adenovirus,is immunogenic. In
the mouse it has been possible to introduce truncated dystrophin constructs
successfully, and these produce proteins which restored the DGC (Yuasa et al, 1998) To try to get around this
problem, researchers have been working on a new generation of
"stealth" adenovirus carriers that few of the original viral genes
and are designed to attract as little attention as possible from the body's
immune system. These vectors are known
as "gutted" vectors. In addition to prolems related to the
antigenicity of the viruses, the dystrophin protein lacking in DMD patients
would have to be present at the sarcolemma at the normal concentration of 2%
total membrane protein, and being “new” would also attract attention from the
immune system. One possible solution is
to use the mini-dystrophin gene. This
mini-gene was discovered in a mild BDM patient (still able to walk at age 60)
(England, et al 1990), however, it is
known that the same mutation can affect different people in very different ways
and it is possible that the same massive deletion may in other patients be dissasterous. As mentioned previously, a better strategy
would be to produce full length utrophin in the muscles of DMD/BMD patients.
Muscle cell culture as an alternative
strategy
Several studies have concluded that it is possible to excise
myoblasts from mice culture, amplify their numbers by tissue culture and
reintroduce the cells to the same animal.
These myoblasts produce muscle tissue with the correct extracellular
matrix support The tissue even becomes
innervated (Irintchev et al, 1998),
this opens up the possiblity of repoulating diseased muscle with genetically
transformed myoblasts. However, a
number of tumours are produced by this process. More hopeful ![]() is the observation that there seems to be
a progenitor muscle cell population present in the bone marrow that circulates
out of the bone marrow and into peripheral muscle via the circulation. This offers the intriguing possibility of
using this system to re-populate the degenerating muscle with cells carrying
the correct or corrected genes (Partridge, 1998).
is the observation that there seems to be
a progenitor muscle cell population present in the bone marrow that circulates
out of the bone marrow and into peripheral muscle via the circulation. This offers the intriguing possibility of
using this system to re-populate the degenerating muscle with cells carrying
the correct or corrected genes (Partridge, 1998).
![]()

![]()


Conclusions
The
MDX mouse is not a suitable model system for DMD/BMD and the MDX:utrn-/-
mice may not be much better. Indeed
since mouse muscle cell up-regulate utrophin enough to continue regeneration,
the mouse may not be a good choice of animal at all. Dystrophic cats, and dogs are available, but are not favourites
for ethical reasosns. Dystrophin is so
large that it is likely to cause an immune reaction, the mini-dystrophin gene
may not be useful in all genetic backgrounds.
The brightest possibility seems at this stage to increase utrophin
expression in the muscle cells. This
may be best achieved by introducing viruses carrying a suitable cDNA, or
perhaps by the introduction of muscle cell pro-generators transformed with
utrophin encoding cDNAs. For the long
term strategy, it may be advisable to offer genetic councelling to those whose
backgrounds pre-dispose them for some of the recessive MDs along with suitable
genetic tests, and for those DMD/BMD patients successfully treated by genetic
intervention.
References
Deconinck , A.E. et al, (1997).
Utrophin-Dystrophin deficient mice as amodel for Duchenne muscular dystrophy. Cell 90;717-727.
England, S.B. et al (1990) Very mild
muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343; 180-182.
Grady, R.M. et al (1997). Skeletal and
cardiac myopathies in mice lacking utrophin and dystrophin : a model for
Duchenne muscular dystrophy. Cell 90;
729-738.
Irintchev, et al, (1998). Ectopic
skeletal muscles derived from myoblasts implanted under the skin. J.Cell Sci. 111; 3287-3297.
Karpati, G. & Carpenter, S. (1986).
Small-calibre skeletal muscle fibers do not suffer deleterious consequences of
dystrophic gene expression. Am.J.Med.Genet.
25; 653-658.
Partidge T(1998) Fantastic voyage of
muscle progenitor cells. Nature Medicine
4; 554-555.
Porter, J.D. et al, (1998). The sparing
of extracolar muscle in dystrophinopathy is lost in mice lacking utrophin and
dystrophin. J.Cell Sci. 111;
1801-1811.
Rafael, J.A., Tinsley, J.M., Potter,
A.C., Deconinck, A.E. & Davies, K.E. (1998). Skeletal muscle-specific
expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nature Genetics 19; 79-82.
Winder, S.J. (1997). The
membrane-cytoskeleton interface: the role of dystrophin and utrophin. J.Muscle Res. Cell Mot. 18;
617-629.
Yuasa, Y. et al, (1998) Effective
restoration of dystrophin-associated proteins invivo by adenovirus-mediated
transfer of truncated dystrophin cDNAs. FEBS
letters, 425 329-336.