Muscular Dystrophy (the
cause)
Many diseases primarily affecting the musculature are known in humans. These are genetic disorders characterised by progressive muscle weakness and wasting in specific muscle groups. Many of the genes whose malfunction causes these diseases are now known, and have in most cases turned out to encode for completely unknown proteins! The proteins are often cytoskeletal and tend to disrupt the connection between the sarcomere and the extracellular matrix, leading to a cycle of muscle degeneration and regeneration which ultimately destroys the tissue. The study of muscular dystrophies promises not only to bring an eventual cure to these devastating diseases but in the shorter term will lead the way for genetic tests for at risk parents. These studies should also continue to provide insights into the biology of muscles and nerves.
Duchenne/Becker Muscular Dystrophy
 It
is estimated that 250 known heritable diseases in humans are X-linked (Stanbury
et al, 1983). Duchenne muscular dystrophy (DMD) and Becker
muscular dystrophy (BMD) are X-linked genetic diseases caused by mutations in
the same genetic locus. They are
amongst the more common genetic diseases to afflict mankind (1 in every 3500
live male births for DMD and 1 in 30,000 for BMD), and certainly the most
common X-linked disease. There is a
wide variation in severity of symptoms due to different mutations but generally
DMD have an early onset (2-5 years) with progressive weakness (Fig1) normally
requiring a wheelchair by 12-13. DMD
kills usually about 20 years old, by respiratory failure and/or cardiac failure
caused by weakness in the intercostal muscles, the diaphragm, and muscles of
the heart. BDM is much less severe,
having an onset of between 5 and 10 years with wheelchairs being resorted to
between 15 and 16 years; life span is much longer than DMD and extremely
variable. One famous individual was
ambulant at 60 years despite an apparently serious deletion (England et al,1990). Some DMD cases suffer
mental retardation.
It
is estimated that 250 known heritable diseases in humans are X-linked (Stanbury
et al, 1983). Duchenne muscular dystrophy (DMD) and Becker
muscular dystrophy (BMD) are X-linked genetic diseases caused by mutations in
the same genetic locus. They are
amongst the more common genetic diseases to afflict mankind (1 in every 3500
live male births for DMD and 1 in 30,000 for BMD), and certainly the most
common X-linked disease. There is a
wide variation in severity of symptoms due to different mutations but generally
DMD have an early onset (2-5 years) with progressive weakness (Fig1) normally
requiring a wheelchair by 12-13. DMD
kills usually about 20 years old, by respiratory failure and/or cardiac failure
caused by weakness in the intercostal muscles, the diaphragm, and muscles of
the heart. BDM is much less severe,
having an onset of between 5 and 10 years with wheelchairs being resorted to
between 15 and 16 years; life span is much longer than DMD and extremely
variable. One famous individual was
ambulant at 60 years despite an apparently serious deletion (England et al,1990). Some DMD cases suffer
mental retardation.
 The
Dystrophin Gene. After a very long
search, the DMD/BMD causing gene was identified by positional cloning (Koenig
et al, 1988). This was a major
triumph! The gene is huge spanning
2,500 kilobases some 0.05% of the entire human genome, a third of the genome of
E.coli and 52 times that of T4
bacteriophage, or 0.5mm long!!. The
gene is complex, encoding mRNA which is multiply spliced and gives rise to at
least six different proteins. The 14kb
complete dystrophin mRNA results from the splicing of 74 exons! The same
mutation of the dystrophin gene can cause a vastly different outcome to the
patient, even within the same family (Bettecker & Müller, 1989).
The
Dystrophin Gene. After a very long
search, the DMD/BMD causing gene was identified by positional cloning (Koenig
et al, 1988). This was a major
triumph! The gene is huge spanning
2,500 kilobases some 0.05% of the entire human genome, a third of the genome of
E.coli and 52 times that of T4
bacteriophage, or 0.5mm long!!. The
gene is complex, encoding mRNA which is multiply spliced and gives rise to at
least six different proteins. The 14kb
complete dystrophin mRNA results from the splicing of 74 exons! The same
mutation of the dystrophin gene can cause a vastly different outcome to the
patient, even within the same family (Bettecker & Müller, 1989).
The MDX mouse has been used as a model for the human disease but the phenotype is weak, only becoming apparent in aged mice. However, this mutant which resides within the mouse dystrophin gene (Ryder-Cook et al, 1988), does not alter the expression of the shorter transcripts (Hoffman et al, 1987). A new mouse mutation in exon 52 disrupts Dp140and Dp260 in addition to dystrophin (Araki et al, 1997). It is hoped that this mouse will provide a better animal model for DMD.
Figure 2.
Dd427 is the full length gene product.
Note that is has 24 internal repeats. Others proteins are produced
from the gene from alternative promotors
Dp427 has a CH domain –like actin binding region at the immediate N
terminus, which is lacking in the other dystrophin gene products. HoweverDp71 and Dp46 have another actin
binding domain at their N termini (Howard et al, 1998), this is like the
actobindin actin-binding protein (Vandekerckhove et al 1990).
The dystrophin repeat consisting of three helices is also shown
(bottom left)

Proteins encoded by the Dystrophin
gene. In muscle, the main gene product is 427 kDa dystrophin and a very similar dystrophin is
expressed in brain from a different promotor, possibly accounting for the
mental retardation observed in with some DMD patients. Dystrophin accounts for 2% of the sacrolemma
protein (Ohlendieck et al, 1991) and as much as 5% of the sarcolemma cytoskeleton
(Ohlendieck & Campbell, 1991). It
was previously reported that dystrophin expression was limited to muscle and
brain, but it is now known that the full length protein is very widely
expressed albeit at very low levels.
The protein is an extended rod shaped molecule and because the
dystrophin repeat is similar to those of spectrin and alpha-actinin, both of
which form anti-parallel coiled coiled dimers, it was assumed that dystrophin
would do so as well. However, it is now
known that dystrophin does not dimerise and exists as a fairly rigid rod with
several more flexible “hinge” regions.
Of the other dystrophin gene products Dp71 is especially abundant in
brain. Dp260 is expressed in retina
(D’Souza et al, 1995) and is required for normal function
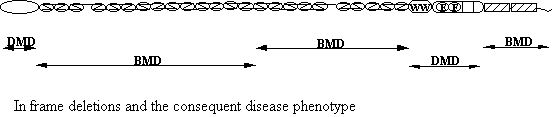
Figure 3
Utrophin. Two years after the sequence of dystrophin was published another highly similar cDNA was reported, this cDNA known initially as dystrophin related protein (DRP) is encoded by chromosome 6 and because of its ubiquitous expression was renamed utrophin.
Other Dystrophin Related Proteins. Dystrobevins are a group of proteins that strongly resemble the C –terminal region of dystrophin. They are encoded by a gene distinct from the dystrophin gene. They are expressed in muscle cells where they co-localise with dystrophin.
 Dystrophin Glycoprotein Complex. The function of dystrophin appears to be to
connect the sarcolemmal cytoskeleton to the extra-cellular matrix. This is accomplished through a series of
interactions with a large group of membranous proteins, the so
Dystrophin Glycoprotein Complex. The function of dystrophin appears to be to
connect the sarcolemmal cytoskeleton to the extra-cellular matrix. This is accomplished through a series of
interactions with a large group of membranous proteins, the so
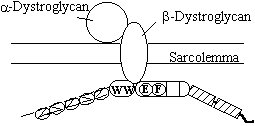
called dystrophin glycoprotein complex (DCG).
The main proteins in this complex are the Dystrogylcans a and b,
these are actually encoded by the same gene and post translationally
cleaved. b-dystroglycan
binds to the C-terminal region of dystrophin (and utrophin) via the WW domain
which recognises the PPPY region of b-dystroglycan. Both the EF hands and the ZZ domain
stabilize this interaction. a-dystroglycan.
Binds the extracellular matrix through interactions with Merosin (a muscle
Laminin) perlecan and agrin (Henry & Campbell, 1999). a-dystroglycan is also known to be the
recepetor used by the Arenaviruses (Lasa fever) and together with Laminin, the
receptor for the bacterium Mycobacterium
leprae (causes Figure 4 leprosy). The dystroglycans are highly conserved
proteins, no disease has yet implicated mutation of the dystroglycan gene in
human disease presumably because it is so widely expressed and has function
other than its role in dystrophin anchoring.
There are a host of other proteins that are thought to regulate and/or
stabilise the dystrophin dystroglycan interaction.
Sarcospan A 25-kDa glycoprotein that spans the membrane four times (Crosbie et al, 1997), that co-localises and co-purifies with the dystrophin-dytroglycan complex.
Sarcoglycans. A group of four transmebranous proteins that are thought to stabilise the DCG (and indeed are thought to be part of it). Mutations in the genes for all four proteins are responsible for some, but by no means all of the LimbGirdle M.D.s (see later Table 1). The sarcoglycans are named a, b, g, and d. The sacroglycans are probably present in the sacrolemma as a complex, as the loss of one member (through mutation) results in the loss of all forms in the membrane (Holt & Campbell, 1998)
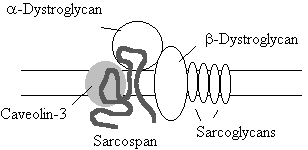
Limb Girdle Muscular Dystrophies
LGMD is a group of muscular dystrophies, which affect the muscles of the hip and shoulder girdles. Diagnosed on this basis. Like DMD, some forms of LGMD are more severe than others. Unlike DMD/BMD, there are many genes and candidate genes involved in LGMD, and the disease has recently been reclassified to reflect this (see table 1). Many (but not all) LGMD result from an absence of functional sarcoglycans components of the dystrophin glycoprotein complex (DCG). Other LGMD result from the absence of functional caveolin-3 (table 1) Calpain-3 binds titin (thin filament) and has a nuclear localization signal. It is thought that the lack of calpain activity in the nucleus may allow a transcription factor to remain active longer than usual.(Ono et al, 1998).
Figure 6 Immunofluorescence staining of a
section of human heart stained with anti-emerin polyclonal
antibody. Emerin is nuclear as well
as present at the intercalated disc, areas of cell-cell adhesion in the
heart.
Other muscle tissues only show nuclear staining.

Gene name |
Gene product |
|
LGMD1A |
? |
|
LGMD1B |
? |
|
LGMD1C |
Caveolin 3 |
|
LGMD2A |
Calpain 3 |
|
LGMD2B |
Dysferlin |
|
LGMD2G |
? |
|
LGMD2H |
? |
|
LGMD2D |
a-Sacroglycan |
|
LGMD2E |
b-Sacroglycan |
|
LGMD2C |
g-Sacroglycan |
|
LGMD2F |
d-Sacroglycan |
Table 1 Figure 6
Emery-Dreifuss Muscular Dystrophies
Emery-Dreifuss Muscular Dystrophy is another X-linked disease, caused by mutations in the gene encoding Emerin. The pathological features of EDMD are similar to DMD/BMD, but emerin is a nuclear protein not found at the sacrolemma. Emerin binds to lamins (not to be confused with Laminins at the cell surface). Lamins are intermediate filaments that are thought to regulate the expression of some genes and so it is possible that Emerin indirectly regulates the expression of a gene (or genes) needed for normal muscular development. It may also be that satalite cells need emerin in order to multiply and differentiate into muscle cells. Anothr possibility is that Emerin is involved in cell-cell adhesion as it has been localised to the intercalated disc in heart (Cartengi et al, 1997).

Myotonic Dystrophy
Myotonic dystrophy (DM) is an autosomal dominant disease that shows an unusual phenomena of ‘anticipation’ where progeny of affected individuals show earlier onset and greater severity of the symptoms. DM is a neuromuscular disorder characterized by wasting of the face and neck muscles and those of the upper body in general. A number of other organs can be affected including the GI tract, testicular atrophy and cardiac conductance problems. More trivial associated problems which may help clinical diagnosis include frontal baldness. The disorder has been linked to the 3' untranslated region of the myotonic protein kinase gene (DMPK), where an expansion of a polymorphic CTG repeat region exists. Interestingly, a number of other human genetic diseases such as Huntington’s disease are also associated with CTG repeat expansions. In DM expansion affects the expression of the DMPK gene and another neighboring gene, DMAHP/SIX 5. The situation is complex and it is not clear if the effect is directly related to the DMPK gene product.
Figure 7. King
Akhenaton a heretical Pharaoh reputedly an DM sufferer (Cattaino et al, 1999 Eur.Neurology 41; 59-63)
Congenital Muscular Dystrophy
Caused by the lack of functional merosin (laminin a2), some symptoms like LGMD. Another form is caused by a lack of integrin a7 perhaps throught the actin binding protein talin (Hayashi et al, 1998).
Fukuyama-type Congenital Muscular Dystrophy
This is one of the most common autosomal recessive disorders in Japan (about 1in 10,000 births). Symptoms include a general muscular dystrophy and mental retardtion caused by the failure of neuronal migration. The responsible gene encodes for a 60kDa secreted protein “Fukutin” (Kobayashi et al, 1998). Uniquely (so far) this is the only human disease caused by an ancient retrotransposon integration which disrupts expression of the fukutin gene. It is thought that this mutation arose once in a single ancestor of the present population. Genetic drift rather than evolution is likely to account for the mutant genes abundance today.
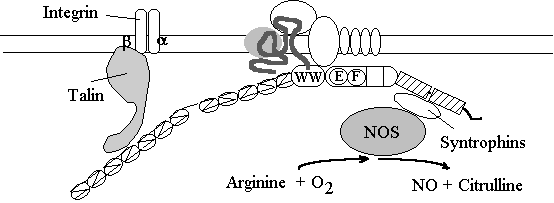
Talin, a very widely expressed actin binding protein protein is also found in muscle cells (at fairly low concentrations). This protein is known to bind Integrins, and integrins have been shown to be mutated in some very unusual and rare cases of MD. This link may not be very important in the mature tissue, but does seem to be important in muscle development. A group of proteins called syntrophins have been found to bind the coiled-coil region of dystrophin. The syntrophin in turn bind Nitric Oxide Synthase. In DMD patients this enzyme is present at low concentration. NO produced by this enzyme decreases contractility and so its loss may cause contractures associated with DMD (Kobzik et al, 1994). It is possible that this connection stops the muscle cell ripping apart!
References
Anderson LVB et al (1999) Dysferlin is a plasma membrane protein and is
expressed early in human development. Hum Mol Gen 8: 855-861
Araki, E. et al (1997). Targeted disruption of exon 52 in the mouse
dystrophin gene induced muscle degenration similar to that observed in Duchenne
muscular dystrophy..Biochem
Biophys.Res.Comm. 238 492-497.
Bashir R, Strachan T, Keers S, Stephenson A, Mahjneh I, Marconi G, Nashef
L & Bushby KMD (1994) A gene for autosomal recessive limb girdle muscular
dystrophy maps to chromosome 2p. Hum Molec Genet. 3: 455-457
Bashir, R, Strachan T, Keers S, Passa-Bueno R, Zatz M, Weissenbach J,
Le Paslier D, Meisler M & Bushby KMD (1996) Genetic and physical mapping at
the limb-girdle muscular dystrophy locus on chromosome 2p. Genomics 33: 46-52
Bettecken, T & Muller, C.R. (1989) Identification of a 220-kb
insertion into the duchenne gene in a family with an atypical course of
muscular dystrophy. Genomics. 4;
592-596.
Bittner et al (1999) Dysferlin deletion in SJL mice defines a natural
model for limb girdle muscular dystrophy type 2B. Nature Genetics 23: 141-142
Bushby K and Beckmann JS. (1995) The limb-girdle muscular dystrophies -
proposal for a new nomenclature. Neuromuscular Disorders 5: 337-344
Bushby K
(1999) Making sense of the limb-girdle muscular dystrophies. Brain
122:1403-1420
Cartegni, L. et al (1997) Heart specific localization of emerin. Hum.Mol.Biol. 13; 2257-2264.
Crosbie R.H.et al, (1997) Sarcospan, the 25-kDa transmebranous
component of the dystrophin-glycoprotein complex. J.Biol.Chem. 272; 31221-31224.
Ellis, J.A., Craxton, M., Yates, J.R.W. & Kendrick-Jones, J. (1998)
Aberrant intracellular targeting and cell-cycle phosphorylation of emerin
contribute to the Emergy-Dreifuss muscular dystrophy phenotype. J.Cell Sci. 111; 781-792.
England, S.B. et al (1990) Very mild muscular dystrophy associated with
the deletion of 46% of dystrophin. Nature
343; 180-182.
D’ouza, V.N. et al (1995). A novel dystrophin isoform is required for
normal retinal electrophysiology. Hum.Mol.Gen. 4; 837-842.
Henry,
M.D. & Campbell, K.P. (1999). Dystroglycan inside and out. Curr.Op.Cell Biol. 11; 602-607.
Holt,
K.H. & Campbell, K.P. (1999). Assembly of the sacroglycan complex. J.Biol.Chem. 273; 34667-34670.
Howard,
P.L. et al (1998) Identification of a novel actin binding site within the Dp71
dystrophin isoform. FEBS letters 441;
337-341.
Kameya, et al (1999). a1-syntrophin gene disruption results in the absence of neuronal-type
nitricoxide synthase at the sarclemma but does not induce muscle degeneration. J.Biol.Chem. 274; 2193-2200.
Kobzik,
et al (1994) Nature 372;
546-548
Kobayashi, K. et al (1998) An ancient retrotransposal insertion causes
Fukuyama-type congentital muscular dystrophy. Nature 394; 388-392.
Koenig, M. Monaca, A.P. & Kunkel, L.M. (1988) The complete sequence
of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53; 219-228.
Ohlendieck,
K., Matsumura, K., Ionasescu, V.V, Towbin, J.A., Posch, E.P., Weinstein, S.L.,
Sernett, S.W. & Cambell, K.P. (1991). Duchenne muscular dystrophy:
deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 43; 795-800.
Ohlendieck, K. & Campbell, K.P. (1991) Dystrophin constitutes 5% of
membrane cytoskeleton in skeletal muscle. FEBS
letters 283; 230-234.
Ono, Y. (1998) Functional defects of a muscle – specific calpain, p94,
casued by mutation associated with Limb-Girdle muscular dystrophy type 2a. J.Biol.Chem. 273; 17073-17078.
Senter,
L., Luise, M., Presotto, C., Betto, R., Teresi, A., Ceoldo, S.
Stanbury, J.B., Wyngaarden, J.B.Fredrickson, D.S., Goldstein, J.L.and
Brown, M.S. (eds) (1983). The Metabolic basis of inhertited disease. P
3-39. McGraw-Hill.
Ryder-Cook, A.S. et al (1988) Localization of the MDX mutation within
the dystrophin gene. EMBO J. 7;
3017-3021.
Winder,
S.J. (1997). The membrane-cytoskeleton interface: the role of dystrophin and
utrophin. J.Muscle Res. Cell Mot. 18;
617-629.